TCR and BCR Analysis
TCR and BCR overview
In combination with other BD Rhapsody™ assays, optional protocols and products enable the generation of sequencing libraries specific to T- and B-Cell Receptors (TCR and BCR). When enabled, the BD Rhapsody™ Sequence Analysis Pipeline can use the reads from these libraries to assemble contigs corresponding to rearranged TCR and BCR chain mRNA. These contigs are then analyzed to identify single-cell level VDJ gene segments, complementary determining regions (CDRs), read and molecule counts, and per cell type chain-pairing. TCR and BCR analysis is supported for human and mouse.
Major TCR and BCR pipeline steps
- Identify reads derived from TCR or BCR mRNA
- Assemble reads into contigs
- Annotate contigs with VDJ gene segment information
- Select dominant contigs and chain family
- Cell analysis - type and quality
- Error correction and contig trimming
Identify reads derived from TCR or BCR mRNA
As described in earlier steps, reads are aligned against a reference sequence to determine their biotype and identity (for example, which gene, AbSeq, sample tag, or VDJ gene segment).
For the WTA assay, in combination with TCR and BCR, the pipeline will identify TCR or BCR reads which align to known VDJ gene segments in the transcriptome, with the appropriate orientation. Known VDJ segments are those with a transcriptome GTF “gene_biotype” starting with “TR_”or “IG_”.
For the targeted assay in combination with TCR and BCR, the pipeline automatically adds species appropriate TCR and BCR gene segments to the FASTA reference file. These gene segments are derived from the same Gencode transcriptome GTF as is used in the pre-built WTA assay reference archives.
Reads that align to TCR or BCR gene segments are grouped and separated from the reads aligning to other biotypes. These reads then only go through the procedures described in the remainder of this section, and not the steps described previously for other biotypes.
Assemble reads into contigs
To generate the full variable region of a TCR or BCR sequence, or the consensus CDR3 sequence, short reads must be assembled. Read assembly operates by looking for similarities and overlaps between reads that suggest they originate from the same original sequence. Aligning and stitching these reads together can allow for the creation of longer contigs from short reads, and correct randomly distributed sequencing errors.
Reads identified as TCR or BCR derived, are prepared for assembly with trimming and UMI error correction. First, the 3’ end of reads are trimmed with a quality score threshold of 20. It’s important that the reads going into assembly be of high quality, so that reads can be correctly aligned, and a valid consensus sequence can be generated. Next, reads are also trimmed based on bead capture sequences to remove artifacts from the BD Rhapsody™ cell label sequence. These sequences could interfere with the correct assembly, and may be found at the 3’ end of TCR or BCR reads if they were derived from short amplicons. Then, reads undergo UMI error correction, grouped by their cell ID and the TCR or BCR chain type determined by initial alignment (for example, TCR-Alpha, IG-Kappa...). This UMI error correction step uses the same RSEC algorithm described previously.
To begin assembly, reads are grouped by their cell ID and chain type. These read groups are sent through a software package for transcript assembly called Trinity, which generates a list of contig sequences. Then, the reads are aligned back to the newly generated contigs, in order to produce read and molecule counts for each contig. Multiple contigs from each cell represent the rearranged VDJ mRNA sequences, for instance, of TCR Alpha and TCR Beta chains.
Annotate contigs with VDJ gene segment information
All contigs generated by the assembly step are analyzed to identify V, D, J, and C gene segments, complementarity determining regions (CDR1-3), framework regions (FR1-4), productivity (lack of stop codons), if contig is full length, and protein sequence. This analysis is accomplished with a software package called IGBlast and with alignments using Bowtie2.
A contig is removed from further analysis if a V or J gene selection is of low quality, indicated by an e-value score greater than 10-3 (lower is better). A contig is considered “full length” when there is amino acid sequence defined for each framework (1-4) and CDR (1-3) region. For the “full length” metric, FR1 and FR4 may be partial, but the overall contig is still considered full length.
All annotated contigs are written to an unfiltered AIRR compliant output file.
Select dominant contigs and chain family
For each cell and chain type, a dominant contig is selected to facilitate reporting, metrics, and downstream analysis. The selection of a dominant contig follows these rules:
-
Read count of the contig is at least 20% of the total read count from all contigs for the cell-chain.
-
To break any ties, the contigs are then sorted in order of: productivity, highest molecule count, highest read count, full length, and best V-segment e-value quality score.
Secondary contigs can be generated due to biological reasons, like dual expression of alpha or beta TCR chains expression or assay-based reasons, like: sequencing errors, transcription errors, contaminating reads from other mRNAs, cell multiplets, and misassembly.
Dominant contigs for chain types that do not correspond to the cell type selected (described below) are not reported in the dominant contigs output file, but all contigs are available in the unfiltered contigs output file.
For cells expressing both TCR alpha/beta and TCR gamma/delta, a single chain family is selected for the final "per cell" output file, but all data is still available in the uncorrected output. To select the chain family, expression of both alpha and beta or gamma and delta is preferred. Then, if all 4 chains or one of each chain has expression, the family with the highest combined molecule count is selected (alpha+beta vs gamma+delta).
Cell analysis - type and quality
During any BD Rhapsody™ assay in combination with TCR and BCR, putative cell determination is still based on 3’ gene expression from targeted or WTA data. This is more accurate than creating separate putative cell identifications for the TCR and/or BCR libraries. The VDJ metrics contain a breakdown of metrics by cell type. Cell types are determined in one of two ways. The pipeline contains an experimental immune cell type classifier that uses a series of machine learning models developed on human PBMCs. This method will only work when the targeted or WTA gene expression, or AbSeq expression data tables contain a sufficient number of core genes/AbSeqs relevant to the model.
The TCR and BCR algorithms contain a simple fallback for cell type determination, in the case of human data where gene expression was not available for a sufficient number of core genes, or for mouse data:
- A putative cell with 2x more TCR molecules than BCR molecules, (or only TCR data) is a T cell
- A putative cell with 2x more BCR molecules than TCR molecules, (or only BCR data) is a B cell
- A putative cell without a 2x difference, or one without any TCR or BCR data is unknown
Cell type determination by the fallback mechanism may be different in unfiltered data vs corrected data.
Cells are then further classified according to their quality. High-quality B or T cells are those that match these criteria:
- Called a B or T cell by the above cell type classification algorithm
- Contains at least 1 productive contig from a cell type appropriate BCR or TCR chain type
- Contains at least 4 molecules from cell type appropriate BCR or TCR chain type(s)
A 'high-quality' column in the perCell and contig output files show whether a cell satisfied these criteria. Then, an additional set of per cell VDJ metrics is computed. These high-quality metrics are similar to other single-cell VDJ products where there is a putative cell determination for VDJ libraries alone, separate from the putative cell determination from associated gene expression libraries.
Error correction and contig trimming
Dominant contigs from putative cells undergo two additional steps before final reporting. First, the 3’ end of contigs are trimmed based on the identified constant region and the known primer sequence it contains. Any assembled sequence 3’ of the primer sequence, within the constant region, is not consequential to the VDJ region, and possibly assembled in error.
To improve specificity, dominant contigs from putative cells undergo a final round of error correction on a per chain type basis: for each of TCR alpha, beta, gamma, delta, and BCR heavy, kappa, and lambda. This distribution style error correction assumes that each individual chain type from T or B cells will amplify at similar rates, and thus would end up with similar numbers of reads per cell-chain for a real TCR or BCR expressing cell. Artifact molecules created with non-T or non-B cell labels in late PCR cycles, such as those derived from PCR chimera formation, will likely have fewer reads. The algorithm is multi-modal aware, so that if there are 2 positive populations for a particular chain, they should both be kept (for example, Naïve B cells and plasma B cells with different reads per cell in the same experiment).
First, there is a check to determine if each chain type has a read depth of at least 4 reads per cell. If not, then error correction does not proceed for that chain type. Next, a histogram of the reads per cell from each chain type is generated, and a multi-modal distribution is modeled on each. A threshold is set at the local minima between the first and second modes, and on a per chain basis, any TCR or BCR data from cells whose reads counts are in the lowest mode are removed.
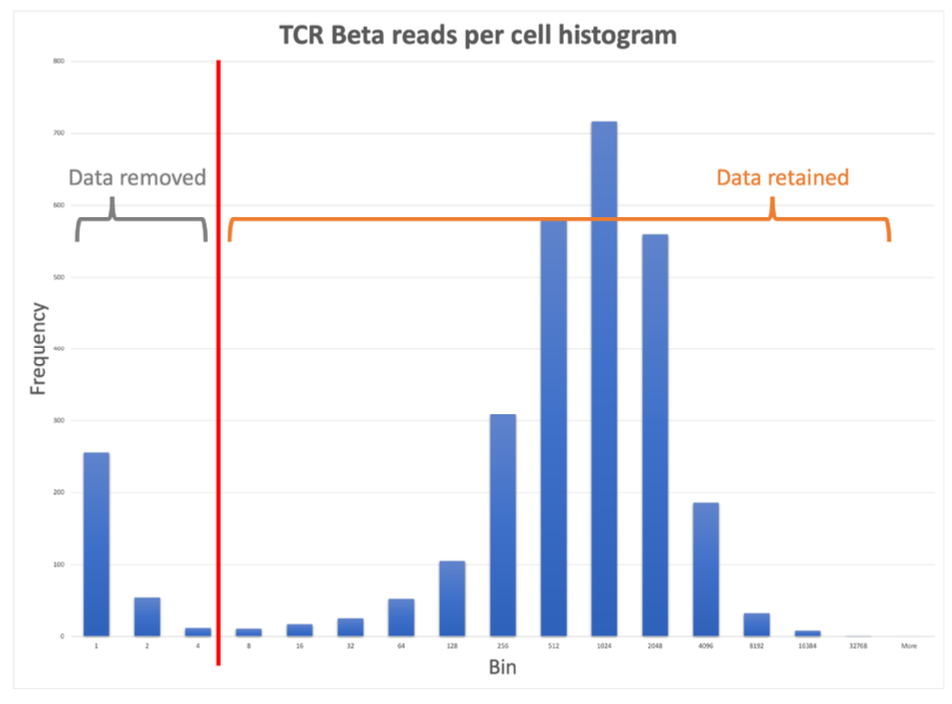
There are two exceptions where contigs will be retained, even if they fall below the threshold for that chain:
- The cell chain contig has a unique CDR3 that is not seen in other cells
- The CDR3 paired data matches exactly to another cell that had both chains pass filter (i.e. there is another alpha-beta T cell clone with the same CDR3 pair)
Untrimmed contigs and contigs before error correction are still available in an unfiltered contigs output file.